


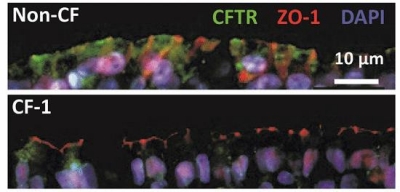
Top panel: Immunofluorescence showing expression and localization of WT-CFTR (green) on primary nasal tissue obtained from a non-CF family member. Tight junction protein ZO-1 (red) and nuclei (DAPI, blue) are also labeled. Bottom panel: Expression and localization of ΔI1234_R1239-CFTR (green) from a family member with CF (i.e. Patient CF-1).
The laboratory of Christine Bear, together with the group of Régis Pomès and collaborators at The Hospital for Sick Children and Proteostasis Therapeutics, used in silico, in vitro and ex vivo techniques to comprehensively understand the consequences of a rare Cystic Fibrosis (CF) disease-causing mutation in the CFTR gene: c.3700 A>G (ΔI1234_R1239), and subsequently develop a novel mechanism-based therapeutic strategy.
Steven Molinski, first author of the study and recent PhD graduate of the Department (March 2016), led the interrogation of this rare variant and showed that it exhibited several protein defects that were comparable to the major mutation, ΔF508; suggesting that drugs designed for ΔF508 (i.e. Orkambi®) may also be effective for this rare variant. However, patient-derived tissue from individuals bearing this rare mutation showed that additional unique defects were present (i.e. low CFTR protein abundance) and that Orkambi® alone would not be sufficient. In collaboration with Proteostasis Therapeutics, they determined that an investigational small molecule “amplifier” (PTI-CH), aimed at enhancing CFTR protein abundance, improved the rescue effect of Orkambi®, first in CRISPR/Cas9-edited bronchial epithelial cells and then in nasal epithelial tissue from individuals bearing this rare mutation. This multi-disciplinary approach provides a novel strategy to treat patients bearing this rare CF-causing mutation, as well as facilitates therapy development and thus personalized medicine for other patients with rare CFTR mutations.
The findings were published in the journal EMBO Molecular Medicine.
{kind=link}